File Formats: Data Tracks
BAM#
To load a set of BAM files merged into a single track see Merged BAM File.
A BAM file (.bam) is the binary version of a SAM file. A SAM file (.sam) is a tab-delimited text file that contains sequence alignment data. These formats are described on the SAM Tools website: http://samtools.github.io/hts-specs/.
BAM, rather than SAM, is the recommended format for IGV. Starting with IGV 2.0.11, IUPAC ambiguity codes in BAM files are supported.
Indexing: IGV requires that both SAM and BAM files be sorted by position and indexed, and that the index files follow a specific naming convention. Specifically, a BAM index file should be named by appending .BAI to the bam file name. A SAM index filename is created by appending .SAI.
-
The index files must have the same base file name and must reside in the same directory as the file that it indexes.
- For example, the index file for test-xyz.bam would be named test-xyz.bam.bai or test-xyz.bai.
-
Multiple tools are available for sorting and indexing BAM files, including igvtools, the samtools package, and in GenePattern. The GenePattern module for sorting and indexing is Picard.SortSam.
- SAM files can be sorted and indexed using igvtools. Note: The .SAI index is an IGV format, and it does not work with samtools or any other application.
Chromosome names: Chromosome names must be consistent between the selected reference genome and the SAM/BAM data files. For convenience, IGV equates chromosome numbers and names of the form chr# (e.g., 1 and chr1 are equivalent).
One-based index: Start and end positions are identified using a one-based index. The end position is included. For example, setting start-end to 1-2 describes two bases, the first and second in the sequence.
BED#
A BED file (.bed) is a tab-delimited text file that defines a feature track. It can have any file extension, but .bed is recommended. The BED file format is described in detail on the UCSC website.
Notes:
IGV does not currently support multiple track lines in a single BED file
Zero-based index: Start and end positions are identified using a zero-based index. The end position is excluded. For example, setting start-end to 1-2 describes exactly one base, the second base in the sequence.
Display settings: To modify IGV's default display settings for the BED data, include a track line in the file.
GFF tag option: By adding a #gffTags line to the beginning of a .bed file, you can add GFF3-style attributes to the Name field (column 4) of a BED file which are displayed in the mouse hover popup text.
- The GFF Name property will become the display name of the feature.
- You must URL encode spaces and other whitespace (e.g. replace space with %20). This is not a requirement of gff3, rather required because bed files are whitespace delimited.
See the GFF3 specification, column 9 for more details.
bedGraph#
The bedGraph format allows display of continuous-valued data in track format. This display type is useful for probability scores and transcriptome data. This track type is similar to the wiggle (WIG) format, but unlike the wiggle format, data exported in the bedGraph format are preserved in their original state. For more information on this file format, see the UCSC website.
Recognized Extension: .bedgraph
BEDPE#
The BEDPE format supports two primary use cases:
- Interactions
- Structural variants
File format variants:
- Original bedtools specification
- Specification for 10X BEDPE files with additional columns. 10X files are recognized in IGV by looking for the following header line:
# chrom1 start1 stop1 chrom2 start2 stop2 name qual strand1 strand2 filters info
Rendering
- Arcs
- Blocks
bigBed#
The bigBed format stores annotation items that can either be simple, or a linked collection of exons, much as BED files do. BigBed files are created initially from BED type files, using the UCSC program bedToBigBed. The resulting bigBed files are in an indexed binary format. The main advantage of the bigBed files is that only the portions of the files needed to display a particular region are transferred, so for large data sets bigBed is considerably faster than regular BED files.
The bigBed format is described in detail on the UCSC website.
bigGenePred#
The bigGenePred format stores positional annotations for collections of exons in a compressed format. bigGenePred files can be created using the UCSC program bedToBigBed.
The bigGenePred format is described in detail on the UCSC website.
bigNarrowPeak#
The bigNarrowPeak format stores called peaks of signal enrichment based on pooled, normalized (interpreted) data. bigNarrowPeak files can be created using the UCSC program bedToBigBed.
The bigNarrowPeak format is described in detail on the UCSC website.
bigWig#
The bigWig format is for display of dense, continuous data that will be displayed as a graph. BigWig files are created initially from WIG type files, using the UCSC program wigToBigWig. Alternatively, bigWig files can be created from bedGraph files, using the UCSC program bedGraphToBigWig. In either case, the resulting bigWig files are in an indexed binary format. The main advantage of the bigWig files is that only the portions of the files needed to display a particular region are transferred, so for large data sets bigWig is considerably faster than regular WIG files.
The bigWig format is described in detail on the UCSC website.
Birdsuite Files#
Birdseye Canary Calls
The file extension must be .birdseye_canary_calls an example file being named:
mycalls.birdseye_canary_calls
The expected file format looks like this:
| sample | sample_index | copy_number | chr | start | end | confidence |
| 1234.CEL | 1 | 2 | 1 | 51598 | 4639285 | 1685 |
| 1235.CEL | 1 | 3 | 1 | 4641859 | 4649979 | 0.37 |
| 1236.CEL | 1 | 2 | 1 | 4653917 | 15359041 | 6038 |
| 1237.CEL | 1 | 3 | 1 | 15361772 | 15362873 | 0 |
| 1238.CEL | 1 | 2 | 1 | 15366497 | 16743865 | 403.13 |
| 1239.CEL | 1 | 3 | 1 | 16758722 | 16808594 | 0.4 |
These files are output when Birdsuite is run so there are no additional steps required for these files to load.
broadPeak#
A broadPeak (.broadPeak) file is used by the ENCODE project to provide called regions of signal enrichment based on pooled, normalized (interpreted) data. It is a BED 6+3 format. See the UCSC website for more details on the broadPeak format.
CBS#
A SEG file (segmented data; .seg or .cbs) is a tab-delimited text file that lists loci and associated numeric values.
See SEG for details.
Chemical Reactivity Probing Profiles#
IGV supports importing chemical reactivity probing profiles from SHAPE or MAP files. After choosing a file to import, the user will be prompted to select the applicable chromosome and optional strand and starting position. IGV will then create a .wig file (WIG format) and load it.
SHAPE format#
The SHAPE format (.shape) is a tab-delimited text file with two columns and no header.
- 1st column: 1-based nucleotide position
- 2nd column: chemical reactivity value, or -999 to indicate positions with no data
Example file:
1 -999.000000
2 -999.000000
3 -999.000000
4 -999.000000
5 -999.000000
6 0.051832
7 -0.668888
8 0.177740
9 -0.136181
10 0.083320
11 -999.000000
12 -999.000000
13 -0.102030
14 -0.056842
15 0.170690
16 0.203813
MAP format#
The MAP format (.map) is output by the SHAPE-MaP software pipeline ShapeMapper. The .map format is identical to the .shape format, with the addition of a third column containing standard error estimates and a fourth column containing the nucleotide sequence. These additional columns are currently ignored by IGV.
Example file:
1 -999.000000 0.000000 G
2 -999.000000 0.000000 G
3 -999.000000 0.000000 T
4 -999.000000 0.000000 C
5 -999.000000 0.000000 T
6 0.051832 0.056355 C
7 -0.668888 0.886105 T
8 0.177740 0.202396 C
9 -0.136181 0.192588 T
10 0.083320 0.079208 G
11 -999.000000 0.000000 G
12 -999.000000 0.000000 T
13 -0.102030 0.144292 T
14 -0.056842 0.210304 A
15 0.170690 0.067038 G
16 0.203813 0.111248 A
CN#
A CN file (.cn) is a tab-delimited text file that contains copy number data. The CN file format is described on the GenePattern website.
Zero-based index: Physical positions are identified using a zero-based index.
Display settings: To modify IGV's default display settings for the CN data, include a track line in the file.
Example: mynah.sorted.cn
Does IGV assume log2(ratio) or absolute values for copy number? IGV looks for the presence of negative numbers. If it finds them, it assumes that the data is log2(tumor/normal). If it does not find negative numbers, it assumes that the values are absolute, with 2 as the center. These assumptions are used to set the heatmap legend; the legend can, however, be changed manually under the View> Color Legends.
For data with negative numbers, IGV defaults to a blue-to-red scale that corresponds to copy numbers from -1.5 to 1.5. Both deletions and amplifications can have continuous valued numbers represented by shading.
CRAM#
CRAM files are used to store aligned sequence data. The specification can be found at http://samtools.github.io/hts-specs/CRAMv3.pdf.
A corresponding index file is required. By convention, the index file name should be the same as the data file name, with “.crai” appended. For example, if the data file is named example_xyz.cram, the index file should be named example_xyz.cram.crai or example_xyz.crai.
genePred#
The genePred table formats can be used to specify the gene track annotations for an imported genome.
Several variations of the genePred table format are described in the FAQ titled “genePred table format” on the UCSC website. Downloading gene information from any of these tables creates a tab-delimited text file where the columns in the file match the columns in the table. Downloaded files may be zipped with a .txt.gz extension. Such a zipped file can be used to specify the gene track annotations for an imported genome. IGV looks for specific string in the filename (case insensitive) to identify the file format:
| File Name Contains | Description |
|---|---|
| ucscGene | Columns in the file match the columns in the table, as described in the “Gene Predictions” section of the genePred table format FAQ. |
|
genePredExt refGene ensGene |
These files have the same format. Columns in the file match the columns in the table, as described in the “Gene Predictions (Extended)” section of the genePred table format FAQ. Note: The first column of this file holds an integer, which is not documented in the FAQ and is ignored by IGV. |
| refFlat | Columns in the file match the columns in the table, as described in the “Gene Predictions and RefSeq Genes with Gene Names” section of the genePred table format UCSC FAQ. |
GFF/GTF#
A General Feature Format (GFF) file is a simple tab-delimited text file for describing genomic features. There are several slightly but significantly different GFF file formats. IGV supports the GFF2, GFF3 and GTF file formats.
- GFF2 files must have a .gff file extension for IGV. See the Wellcome Trust Sanger Institute website (https://ensembl.org/info/website/upload/gff.html) for a description of the GFF2 file format.
- GFF3 files must have a .gff3 file extension for IGV. See the Sequence Ontology Project (SO) website (http://www.sequenceontology.org/gff3.shtml) for a description of the GFF3 file format.
- GTF files must have a .gtf file extension for IGV. See the Computational Genomics Laboratory website (http://mblab.wustl.edu/GTF2.html) for a description of the GTF file format.
One-based index: Start and end positions are identified using a one-based index. The end position is included. For example, setting start-end to 1-2 describes two bases, the first and second in the sequence.
Display settings: To modify IGV's default display settings for the .gff or .gff3 data, include a track line in the file.
Feature display name: To override the default setting for which field is used to label the features in the IGV track, add the following line to the file:
##displayName=<field name>
Coloring features: To specify a color for a given feature, you can add this to the file as shown in the following example. Color values can be in either hexadecimal or RGB (r, g, b) format.
##gff-version 3
chr1 varclass variants\_454HCDiffs 59133 59133 33 . . Var=A->G;AA=S->S;depth=9;frame=+1;gene=OR4F5;ref=novel;InRegion;color=#0000EE
chr1 varclass variants\_454HCDiffs 59374 59374 67 . . Var=A->G;AA=T->A;depth=30;frame=+1;gene=OR4F5;ref=rs2691305;InRegion;color=#EE0000
chr1 varclass variants\_454HCDiffs 731442 731442 100 . . Var=T->C;AA=->;depth=3;frame=;gene=;ref=rs3115865,rs61770168;OutOfRegion;color=#AAAAAA
GISTIC#
A GISTIC file (.gistic) is the Gistic Scores File output from the GenePattern GISTIC module. It is a tab-delimited text file that defines a feature track displaying the q-value for regions of amplification or deletion found using GISTIC (Beroukhim et al., 2007). The first row contains eight column headings, which must be identical to those listed in the following table. Each subsequent row defines a GISTIC feature.
IGV displays GISTIC deletion scores as a blue line and amplification scores as a red line:
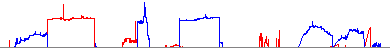
Example: scores.gistic
| Column Heading | Description |
|---|---|
| Type | Aberration type, which is specified as Amp or Del (amplification or deletion) |
| Chromosome(hg17) | Chromosome |
| Start | Location of the first base pair in the aberrant region |
| End | Location of the last base pair in the aberrant region |
| q-value | False Discovery Rate q-values for the aberrant regions (q-values below a user-defined threshold are considered significant) |
| score | G-score that considers the amplitude of the aberration as well as the frequency of its occurrence across samples |
| amplitude | Average amplitudes among aberrant samples |
| frequency | Frequency of aberration across the genome for both amplifications and deletions |
GWAS#
A GWAS file is a space- or tab-delimited result file from genome-wide association study (GWAS) analysis. These files include PLINK result files containing integrated map information (i.e., chromosomal location for each association).
File extensions for GWAS files are: .linear, .logistic, .assoc, .qassoc, .gwas
GWAS file must contain a header line and four required columns (case-insensitive):
- CHR: chromosome (aliases chr, chromosome)
- BP: nucleotide location (aliases bp, pos, position)
- SNP: SNP identifier (aliases snp, rs, rsid, rsnum, id, marker, markername)
- P: p-value for the association (aliases p, pval, p-value, pvalue, p.value)
Columns can be in any order. Other columns besides the required ones are allowed and will be included in popup text. The p-value will be transformed to -log10 scale for plotting.
IGV#
An IGV file (.igv) is a tab-delimited text file that defines tracks. The first row contains column headings for chromosome, start location, end location, and feature followed by the name of each track defined in the .igv file. Each subsequent row contains a locus and the associated numeric values for each track. IGV interprets the first four columns as chromosome, start location, end location, and feature name regardless of the column headings in the file. IGV uses the column headings for the fifth and subsequent columns as track names. Feature names are not displayed in IGV.
For example:
| Chromosome | Start | End | Feature | Patient-One | Patient-Two | Patient-Three |
| chr1 | 2150459 | 2150460 | Test_one | 0.01 | 0 | 0.99 |
| chr1 | 3558044 | 3558045 | Test_two | 0.25 | 0.71 | 1.31 |
Zero-based index: Start and end positions are identified using a zero-based index. The end position is excluded. For example, setting start-end to 1-2 describes exactly one base, the second base in the sequence.
Data must be grouped by chromosome and within each chromosome group sorted by start position: igvtools can be used to sort .igv files.
Display settings: IGV displays IGV file data using default display settings. To modify the default display settings for the data, you can:
- Include a type line in the file to make IGV use the display settings for a different data type.
- Include a track line in the file.
Custom columns
IGV supports custom specification of columns for the ".igv" file format. To use this, include a column specifier directive at the head of the file. The column directive line starts with #columns, followed by one or more column specifiers of the form key=value. Valid keys are listed in the following table. Columns are tab delimited.
| Key | Value |
|---|---|
| chr | index of the chromosome column (required) |
| start | index of the start position column (required) |
| end | index of the end position column (optional) |
| probe | index of a probe or description column (optional) |
| data | either a single index, or a range in the form of 5-10, of the data columns (required) |
If a single value is entered for the data column, it is interpreted as the "first" data column. All columns starting with this value are assumed to contain data. To specify exactly one column, use a range (e.g., 5-5) to specify the 5th column.
Example:
#columns chr=7 start=8 probe=2 data=4-5 #coords=1
> Index TargetID ProbeID\_A sample\_1\_methylation sample\_2\_methylation genome\_build chromosome position
> 60 cg00002593 25796427 0.7642099 0.7426524 37 1 1258656
> 21 cg00000957 65648367 0.8172337 0.8323303 37 1 5859840
> ....
LOH#
An LOH file (.loh) is a copy number file that contains "loss of heterozygosity" values. The format is identical to the CN format, but the numbers have the following meanings:
- -1: Conflict (homozygous in the normal and heterozygous in the tumor)
- 0: Retained
- 1: Loss of heterozygosity
Numbers that fall between these values represent the probability of LOH. IGV treats the values as a continuum and colors them according to the heatmap scale set for the LOH track.
Display settings: To modify IGV's default display settings for the LOH data, include a track line in the file.
MAF (Multiple Alignment Format)#
The Multiple Alignment Format stores a series of multiple alignments. See the UCSC website for more details. The extension must be ".maf".
.maf files must be in plain text (not gzipped). The alignment blocks in the file must be sorted by start position, and the file requires an accompanying index. If no index file is detected, IGV will create the index when the file is first loaded, which may result in a delay in loading, depending on the size of the file. Do not close IGV while indexing is in progress.
MAF (Mutation Annotation Format)#
A Mutation Annotation Format (MAF) file (.maf) is a tab-delimited text file that lists mutations. The format is described in detail at the NCI's Genomic Data Commons documentation site here.
Merged BAM File#
A set of BAM files can now be loaded merged into a single track.
Create a plain text file containing a list of the BAM files you want to load, listed by either file path or URL. The paths or URLs can be either absolute or relative to the location of the list file. . IGV will load all the BAM files as a single track. The filename must end with the compound extension ".bam.list".
Example:
gs://genomics-public-data/platinum-genomes/bam/NA12889\_S1.bam
gs://genomics-public-data/platinum-genomes/bam/NA12877\_S1.bam
gs://genomics-public-data/platinum-genomes/bam/NA12878\_S1.bam
narrowPeak#
A narrowPeak (.narrowPeak) file is used by the ENCODE project to provide called peaks of signal enrichment based on pooled, normalized (interpreted) data. It is a BED 6+4 format. See the UCSC website for more detail on this format.
PSL#
A PSL file (.psl) is a tab-delimited text file that represents alignments, and are typically taken from files generated by BLAT or psLayout. The PSL file format is described on the UCSC website.
RNA Secondary Structure Formats#
BP (RNA base pairing)#
A BP file (.bp) is text file format that describes connections between ranges of nucleotides, and is primarily used to indicate base pairing interactions or estimated pairing probabilities for RNA structures. BP files are rendered in IGV using colored semicircular arcs.
File Header. A file begins with any number of header lines listing all arc colors and associated labels. Each of these lines are tab-delimited, and must begin with "color", followed by the red, green, and blue color components 0-255, followed by an optional text label which will be shown in the track menu color legend. Arc colors will be rendered in listed order (i.e. the last listed color will be drawn on top). Track lines are not currently supported for this file type.
Example header line: color: 51 114 38 High-probability base pairs
Paired Ranges. Each tab-delimited line in the rest of the file describes a single arc. The first field is the name of the associated IGV chromosome. The last field is a zero-based integer index indicating the arc color (from the colors listed in the header). The second through fifth fields are the 1-based inclusive nucleotide coordinates of paired ranges (a helix, if this is an RNA structure).
Example BP file: example.bp
Additional RNA secondary structure formats can be imported into IGV and converted to the BP format. They include the DB, CT, and DP formats, which are described below. After choosing a file to import, the user will be prompted to select the applicable chromosome and optional strand and starting position. IGV will then create a .bp file and load it.
DB (dot bracket)#
DB (dot bracket) format (.db, .dbn) is a plain text format that can encode secondary structure. Lines beginning with > or # are currently ignored. Nucleotide sequence is currently ignored.
Secondary structure notation:
- Unpaired nucleotides are indicated with the . or : characters.
- Matching pairs of parentheses indicate base pairs.
- To indicate non-nested base pairs (pseudoknots), additional brackets may be used: [], {}, or <>.
Files containing multiple sequences or structures are currently not supported.
Example:
GGUGCAUGCCGAGGGGCGGUUGGCCUCGUAAAAAGCCGCAAAAAAUAGCAUGUAGUACC ((((((((((((((.[[[[[[..))))).....]]]]]]........)))))...))))
CT (connectivity table)#
The CT format (.ct) is used by software packages such as RNAstructure. See the CT File Format on the Mathews Lab web page.
Only the first structure in a CT file will be imported by IGV. CT files with additional headers (often starting with the # character) are currently not supported.
Example CT file: example.ct
DP (dot plot or pairing probability)#
The DP file format (.dp) can be generated using the RNAstructure software package by running partition followed by ProbabilityPlot on the resulting .pfs file with the -t option for text file output. For modeling the structures of large mRNAs, the program Superfold runs partition on multiple overlapping windows, then heuristically merges the windows. Superfold outputs a merged .dp file by default.
File format:
- 1st line is the number of entries in the file.
- 2nd line is column names.
- Remaining lines describe pairing probabilities between 1-based nucleotide positions, given as tab-separated
Upon import, IGV colors pairs above 80% probability dark green. Pairs between 30 and 80% probability are colored blue. Pairs between 10 and 30% probability are colored light yellow.
BED#
IGV also supports viewing RNA secondary structures in BED format. The file must include a track line which species graphType=arc. Each record line must contain the first three columns of a bed file: chrom, start and end, where the start and end represent the base pair. Note that the start position follows standard BED file convention and is zero-based (first base on asequence is position 0).
The following small example represent a hypothetical stem loop:
track graphType=arc
chr1 10 25 stemloop1
chr1 11 24 stemloop1
chr1 12 23 stemloop1
chr1 13 22 stemloop1
chr1 14 21 stemloop1
chr1 15 20 stemloop1
Additional examples can be found in the supplement of the following paper
SAM#
For detailed specifications, we refer you to the September 2014 article titled Sequence Alignment/Map Format Specification by the SAM/BAM Format Specification Working Group, and the Samtools site.
For information on the related binary version of SAM, see BAM.
The citation for the 2009 Bioinformatics paper introducing the SAM format follows:
Li H.*, Handsaker B.*, Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and 1000 Genome Project Data Processing Subgroup (2009) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics, 25, 2078-9. [PMID: 19505943]
SEG#
A SEG file (segmented data; .seg or .cbs) is a tab-delimited text file that lists loci and associated numeric values. The first row contains column headings, and each subsequent row contains a locus and an associated numeric value. IGV ignores the column headings. It reads the first four columns as track name, chromosome, start location, and end location. It reads the last column as the numeric value for that locus (if the value is non-numeric, IGV ignores the row). IGV ignores all other columns.
The segmented data file format is the output of the Circular Binary Segmentation algorithm (Olshen et al., 2004).
Example: example.seg
Display settings: SEG files can include a type line to set display settings.
TDF#
A tiled data file (TDF) file (.tdf) is a binary file that contains data that has been preprocessed for faster display in IGV.
Generate TDF files by using the igvtools toTDF command.
More detailed information about the TDF file format is available here.
Track Lines#
When IGV loads a data file, it uses the file extension to determine the file format, the file format to determine the data type, and the data type to determine the default display options. Adding a track line to a data file modifies IGV's default display options. This can be particularly useful for file formats not associated with any particular type of data, such as the IGV file format.
The following file formats allow track lines:
- BED, WIG, PSL
- IGV, CN, SNP, GFF, LOH, GFF3, SEG -- in these file formats, the track line must begin with a # symbol; i.e. #track
IGV track lines are based on WIG track lines. See the UCSC website for the WIG track line syntax. The following table describes the track line specifiers that IGV supports. IGV includes a few options that are not part of the UCSC specification.
IGV does not support multiple track lines in a single file.
| Specifier | Value | Description |
|---|---|---|
| name | trackLabel | Track name (ignored when used in the IGV file format) |
| description | centerlabel | Currently ignored |
| visibility | full | dense | hide | Currently ignored |
| color | RRR,GGG,BBB | Color for positive values in all tracks |
|
altColor |
RRR,GGG,BBB | Color for negative values in all tracks |
| priority | N | Currently ignored |
| autoScale | on|off | Currently ignored. All tracks autoscale unless an explicit data range is defined (e.g., by including the viewlimits specifier). |
| gridDefault | on | off | Currently ignored |
| maxHeightPixels | max:default:min |
default and min are supported max is currently ignored |
| graphType | bar | points | heatmap |
Graph type to use: chart | scatter plot | heatmap. (IGV only: The heatmap value is an IGV addition to the specification.) |
| midRange (IGV extension) | x:y |
Defines the neutral range for a 3-color heatmap. Values in this range are rendered with the midColor value, which is white by default. Example: midRange=20:80 |
| midColor (IGV extension) | RRR,GGG,BBB |
Color to use in the "mid range" of a heatmap. Example: midColor=0,0,150 |
| viewLimits | lower:upper | Defines the data range |
| yLineMark | real-value | Currently ignored |
| yLineOnOff | on | off | Currently ignored |
| windowingFunction | maximum | minimum | mean | none | Function that summarizes the values in a window of data represented by one pixel |
| smoothingWindow | off | [2-16] | Currently ignored |
| url | Defines a URL for an external link associated with this track. Any '$$' in this string this will be substituted with the item ID if explicitly defined, or name if ID is not specified.. | |
| coords (IGV extension) | 0 | 1 |
Indicate whether the file uses 0 or 1 based coordinates. The UCSC specification for WIG files uses 1 based coordinates and for BED files uses 0 based coordinates. If data looks off by one, check for a possible 0 vs 1 based coordinate issue. |
| scaleType (IGV only) | log | linear | The Y-axis scale type for charts |
| featureVisibilityWindow (IGV only) |
integer value |
The window size in bp below which features are loaded and displayed. When the viewing window is above this value a message is displayed "Zoom in to view features". This parameter is useful for large indexed feature tracks. A negative value indicate features should be loaded for an entire chromosome (but not the whole genome) |
| gffTags (IGV extension) | off | on | If "on" the name field is treated as a GFF3 style attribute list (column 9 of GFF3). The default is "off". |
Type Lines#
When IGV loads a data file, it uses the file extension to determine the file format, the file format to determine the data type, and the data type to determine the default display options. In the IGV and segmented (SEG, CBS) file formats, you can use a #type line to override the default data type and thus the default display options. For example, the IGV file format has a default data type of 'Other' and, therefore, the data in file is displayed using a blue bar chart with an autoscaled data range. By adding a #type line to the IGV file, you can indicate that the file contains gene expression data; in which case, the data will be displayed using a blue-to-red heatmap with the data range set from -1.5 to 1.5.
The type line must be the first line in the file. It has the following format: #type=<data-type>
where <data-type> is one of the following (these values are case-sensitive):
COPY_NUMBER
GENE_EXPRESSION
CHIP
DNA_METHYLATION
ALLELE_SPECIFIC_COPY_NUMBER
LOH
RNAI
The selected data type determines the display settings.
VCF#
VCF, which stands for Variant Call Format, is a standardized text file format used for representing SNP, indel, and structural variation calls. The full specification of the format can be found at https://samtools.github.io/hts-specs. IGV supports VCF version 4.
Required Extensions: .vcf, .vcf.gz
Indexing:
VCF files are not required to be indexed, but the whole file will then be loaded into IGV memory, which is not recommended for larger files.
VCF files can be indexed with igvtools or Tabix:
-
igvtools can be run from the command line or IGV itself (Tools > Run igvtools...) After launching, choose the Index command and browse to your .vcf file. The index file (.idx) will be created in the same directory as the .vcf file. igvtools also sorts .vcf files.
-
Tabix is used to index gzipped files (ending with .vcf.gz); it creates a .tbi file. Tabix, including documentation, is available from the SamTools website.
Example V.4.0 File:
##fileformat=VCFv4.0
##fileDate=20090805
##source=myImputationProgramV3.1
##reference=1000GenomesPilot-NCBI36
##phasing=partial
##INFO=<ID=NS,Number=1,Type=Integer,Description="Number of Samples With Data">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
##INFO=<ID=AF,Number=.,Type=Float,Description="Allele Frequency">
##INFO=<ID=AA,Number=1,Type=String,Description="Ancestral Allele">
##INFO=<ID=DB,Number=0,Type=Flag,Description="dbSNP membership, build 129">
##INFO=<ID=H2,Number=0,Type=Flag,Description="HapMap2 membership">
##FILTER=<ID=q10,Description="Quality below 10">
##FILTER=<ID=s50,Description="Less than 50% of samples have data">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth">
##FORMAT=<ID=HQ,Number=2,Type=Integer,Description="Haplotype Quality">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NA00001 NA00002 NA00003
20 14370 rs6054257 G A 29 PASS NS=3;DP=14;AF=0.5;DB;H2 GT:GQ:DP:HQ 0|0:48:1:51,51 1|0:48:8:51,51 1/1:43:5:.,.
20 17330 . T A 3 q10 NS=3;DP=11;AF=0.017 GT:GQ:DP:HQ 0|0:49:3:58,50 0|1:3:5:65,3 0/0:41:3
20 1110696 rs6040355 A G,T 67 PASS NS=2;DP=10;AF=0.333,0.667;AA=T;DB GT:GQ:DP:HQ 1|2:21:6:23,27 2|1:2:0:18,2 2/2:35:4
20 1230237 . T . 47 PASS NS=3;DP=13;AA=T GT:GQ:DP:HQ 0|0:54:7:56,60 0|0:48:4:51,51 0/0:61:2
20 1234567 microsat1 GTCT G,GTACT 50 PASS NS=3;DP=9;AA=G GT:GQ:DP 0/1:35:4 0/2:17:2 1/1:40:3
This example shows in order:
- A good, simple SNP
- A possible SNP that has been filtered out because its quality is below 10
- A site at which two alternate alleles are called, with one of them (T) being ancestral (possibly a reference sequencing error)
- A site that is called monomorphic reference (i.e., with no alternate alleles),
- A microsatellite with two alternative alleles, one a deletion of 3 bases (TCT), and the other an insertion of one base (A).
Genotype data are given for three samples, two of which are phased and the third unphased, with per sample genotype quality, depth, and haplotype qualities (the latter only for the phased samples) given as well as the genotypes. The microsatellite calls are unphased.
WIG#
A WIG file (.wig) is a text file that defines either a feature or data track. The WIG file format is described on the UCSC website.
Required extension .wig
For faster loading, convert WIG files to bigWig format. Alternatively, convert to TDF format using igvtools.
IGV does not currently support multiple track lines in a single WIG file
One-based index: Start and end positions (for "fixedStep" and "variableStep" formats) are identified using a one-based index. The end position is excluded. For example, setting start-end to 1-2 describes exactly one base, the first base in the sequence.
Display settings: To modify IGV's default display settings for the WIG file data, include a track line in the file.